
Narcolepsy is a debilitating neurological disease affecting around 0.05% of the human population, men and women alike.[1][2] The disease is defined as the inability to regulate sleep/wakefulness states properly. Its primary symptoms are excessive daytime sleepiness (EDS) and cataplexy.
EDS is characterized by a general feeling of sleepiness throughout the day with occasional “sleep attacks” and loss of consciousness. Cataplexy consists of sudden attacks of bilateral skeletal muscle weakness without impairment of consciousness or memory. This is often triggered by emotional reactions.[1] Other less common symptoms are sleep paralysis and hypnagogic hallucinations.[3] These four symptoms combined are known as the “tetrad of narcolepsy” and is present in only about 15% of patients.[2]
Although narcolepsy mainly shows during wakefulness during day-time, 95% of narcoleptic patients have disturbed night-time sleep as well. This includes prolonged non-rapid eye movement (NREM) sleep/rapid eye movement (REM) sleep ultradian cycle, sleep fragmentation, and REM sleep dissociation events.[1][4] Narcolepsy is clinically categorized as occurring with cataplexy, without cataplexy, or secondary to another medical disorder. [3]
To diagnose a patient, usually, polysomnography is used both at night and during a multiple sleep latency test (MSLT). In the test, if REMS occurs in less than 15 min, the epoch is defined as a sleep-onset REMS period (SOREMSP). A mean sleep latency of less than 8 min and 2 or more SOREMSP after minimally sufficient sleep on the previous night indicates narcolepsy. [3]
Causation and treatment
The cause of the disease is most likely the degeneration of hypothalamic neurons that contain the neuropeptide orexin (or hypocretin), produced mainly in the perifornical (lateral) hypothalamus (PFH). Two isoforms (orexin-A and -B) of orexin, derived from proteolytic cleavage of prepro-orexin, exert their actions through two types of G-protein-coupled receptors (OxR1 and OxR2). [1]
The role of orexins and its receptors in narcolepsy was first discovered in studies of narcoleptic dogs and orexin-deficient mice. It was found to play an important role in regulating sleep/wakefulness states, energy homeostasis, metabolism, and addiction. [3][5] Since then, scientists have been able to successfully engineer rodent models of narcolepsy. [1] Human narcolepsy is caused by the loss of about 90% of orexin neurons together with loss of dynorphin and activity-regulated pentraxin (NARP), responsible for sending signals from those neurons. [1][3] The onset of narcolepsy seems to occur typically postpubertal, and mostly in individuals with detrimental environmental exposure and who are susceptible to genetical modifications. [1][5] Moreover, most narcoleptic patients with cataplexy are positive for a specific MHC class II allele, HLA-DQB1*0602. [1][3][4]
Currently, there is no cure for narcolepsy. However, some treatments are available of which the administration of sodium oxybate (the sodium salt of gamma-hydroxybutyrate, GHB) is a leading treatment option, as it treats excessive daytime sleepiness (EDS), cataplexy and disturbed nocturnal sleep. [1] However, more pharmacological research is needed.
Research techniques for studying narcolepsy
Narcoleptic mice make for the most convenient research subject for studying narcolepsy due to their similarities to humans in terms of their genetics, physiology, and anatomy and due to their easy-to-manipulate genome. Thanks to the discovery of the malfunction of the orexin neuron system contributing to narcolepsy and subsequent engineering of different mouse models to mimic orexin loss, mice were created that show similar narcoleptic symptoms to humans. [1] When desired transgenic mouse lines are engineered, it is important that the mice are provided with food and water ad libitum, housed with controlled temperature and humidity, and kept on a 12 hour light/dark cycle at all times. [2]
Behavioral testing
Narcoleptic episodes can be behaviorally scored, in both light and dark periods, by observers that are blinded to the genotypes. The following behaviors are typically scored as cataplectic behavior:
- An abrupt end to clear purposeful motor activity,
- A sustained change in posture maintained throughout the episode,
- An abrupt end to the episode with the continuation of obvious purposeful motor activity.
To put it simply, it seems like a switch turns off and back on in the mouse. Criteria for sleep attacks or gradual arrests are identical except for a more gradual onset (more than 2 sec).[2][6]
The most frequently used test for studying narcolepsy is the open field test. Open field apparatus mounted with an overhead infrared video camera (for night-time activity) can be used to easily measure locomotor activity and score narcoleptic episodes.[2][7] Other tests include food-elicited cataplexy test where chocolate is presented in an open field maze.
Infrared video recordings/photography
Infrared videotaping/photography and digital time recording can be used for documentation and scoring of dark cycle behavior, as it can detect locomotor activity.[2][6][8]
Spontaneous Motor Activity
Spontaneous motor activity can be measured by systems with sensors that continuously measure radiated body heat of the mice[6] or with a weight sensor positioned under the cage.[5]
Electroencephalography and electromyography recordings
The most important tools for measuring narcoleptic behavior are EEG/EMG recordings for polysomnographic assessment. They are used to study the brain-waves of mice models during narcoleptic episodes. Lightweight implants and cabling procedure is used to allow full freedom of movement. EEG/EMG records can be scored into epochs of Awake, REM, and non-REM sleep according to standard criteria of rodent sleep.[2][6] An episode of cataplexy can be defined as an abnormal transition from active wake to a sudden loss of activity: characterized by a period of at least 10 sec of EEG theta activity accompanied by EMG atonia. [5][8]
Phenotypes of Narcolepsy in humans vs. mice
In short, the prominent characteristics of human narcolepsy are, during day-time, sudden attacks of muscle weakness without losing consciousness (cataplexy), extreme feelings of sleepiness (EDS) sometimes accompanied with sleep attacks paired with losing consciousness, and during night-time, prolonged NREM/REM sleep ultradian cycles, sleep fragmentation and a more direct transition from wakefulness to REM-sleep.
In narcoleptic rodent models, these features translate as following phenotypes: sudden behavioral arrests during the dark phase (when mice are normally most active), occurring at the timing of direct transitions from wakefulness to REM sleep, similar to human cataplexy, gradual behavioral arrests during the dark phase, similar to human sleep attacks, and overall fragmented sleep-wake cycles. The latter mentioned can be light or severe depending on the model.[1][4]
Human cataplexy is often triggered by strong emotions, whereas the sudden behavioral arrests in narcoleptic mice are often triggered by palatable foods such as chocolate, purposeful motor activity such as grooming, burrowing, and climbing and social interaction. In juvenile mice, it can be triggered by playful “laughter”, i.e. displaying 50 Hz ultrasonic vocalizations. Wheel running and group-housing seem to increase the amount of cataplexy.[1][3][4] During the episodes of arrest, mice exhibit EEG/EMG patterns similar to those during REMS or NREMS with sleep spindles.[4]
Gradual arrests begin during quiet wakefulness and could be easily distinguished from normal resting behavior by lack of stereotypic preparation for sleep (e.g., nesting) and characteristic “nodding” of the head over a period of several seconds, followed by a collapsed posture. They are not associated with strong emotions or muscle atonia. [4]
Mouse models of narcolepsy
The Orexin−/− mouse
Orexin knockout (KO) models are engineered by disrupting the prepro-orexin gene in order to eliminate orexin ligands: i.e. orexin-A ligands with high affinity for orexin receptor type 1 (OX1R) and orexin-B ligands with more affinity for orexin receptor type 2 (OX2R). OX2R has equal amounts of affinity for both orexin-A and orexin-B. These ligands are needed for energy homeostasis and sleep-wake regulations.[2] Read Chemelli et al. (1999) for a more detailed description of the generation of the mouse lines.[1]
During the dark period, they show frequent periods of sudden behavioral arrests. Moreover, much like human narcolepsy, they show increased REM-sleep time and duration, severe fragmentation of sleep/wakefulness states, and more direct transitions to REM sleep. In addition to abrupt arrests, there can occur other types of gradual arrests. A study conducted by Willie et al. (2003) found that caffeine suppressed these gradual arrests while clomipramine suppressed abrupt arrests, which again shows how similar these arrests are to human cataplexy.[1][9] During the light period, orexin KO mice only show a mild decrease in food intake and have a mild tendency for obesity depending on their genetic background.[1]
The benefits of using this model are that it is easy to replicate due to many studies describing the production of these mice in detail and it shows similar cataplexic attacks to humans, which makes it interesting to study cataplexic behavior. However, unlike in most human cases, orexin neurons are still intact despite the loss of orexin ligands, which makes it a less accurate model. [1][3]
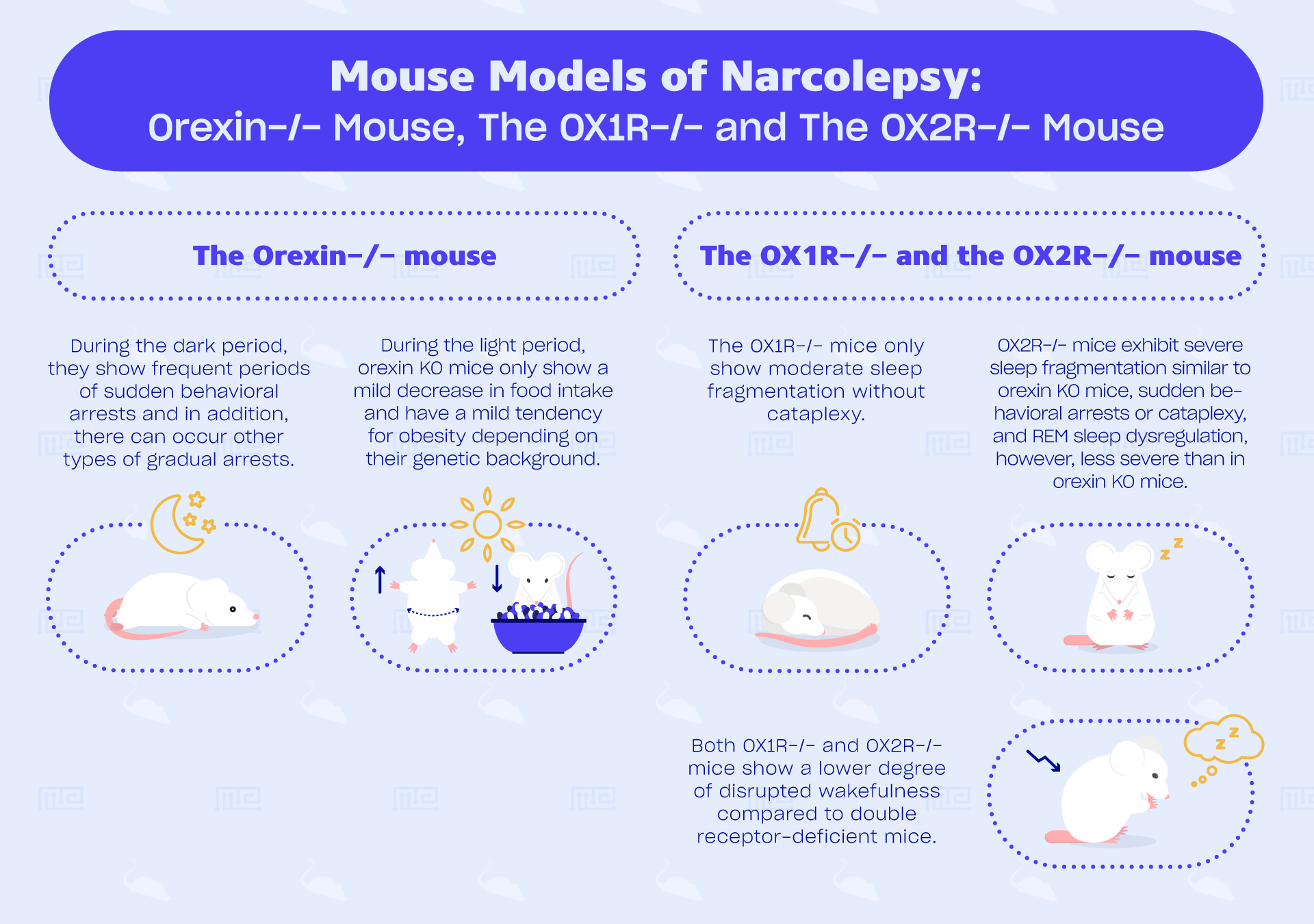
The OX1R−/− and the OX2R−/− mouse
These single receptor KO mice can be created by the mating of heterozygous OX1R+/− mice (for OX1R−/− mice) and of OX2R+/− mice (for OX2R−/− mice) and PCR genotyping. Read Abbas et al. (2015) and Willie et al. (2003) for a more detailed description.[7][9]
OX1R−/− mice only show moderate sleep fragmentation without cataplexy.[1][4] In contrast, OX2R−/− mice do exhibit sudden behavioral arrests, or cataplexy, and REM sleep dysregulation, however, less severe than in orexin KO mice. It was found that its frequency is much less (31 times lower in frequency) and more gradual arrests are observed. Both OX1R−/− and OX2R−/− mice show a lower degree of disrupted wakefulness compared to double receptor-deficient mice.[1]
OX2R−/− mice show severe sleep fragmentation similar to orexin KO mice.[4] By comparing symptoms of orexin KO mice and OX2R−/− mice, the study of Willie et al. (2013) found that orexin regulation of maintaining wakefulness and of normal gating of non-REM sleep requires intact OX2R signaling. The creation of agonists of each of the orexin receptors could provide selective therapeutic benefits in treating specific symptoms of narcolepsy.[9]
In summary, these two single receptor KO models are interesting for studying the specific function of OX1R and OX2R receptors separately. OX1R−/− mice are less interesting when studying human narcolepsy due to its lack of translational validity. OX2R−/− mice show similar phenotypes of narcolepsy as orexin KO mice, however, its cataplectic symptoms are low in frequency. Both models are important for studies investigating their function, as it might contribute to the therapeutic treatments of specific symptoms.
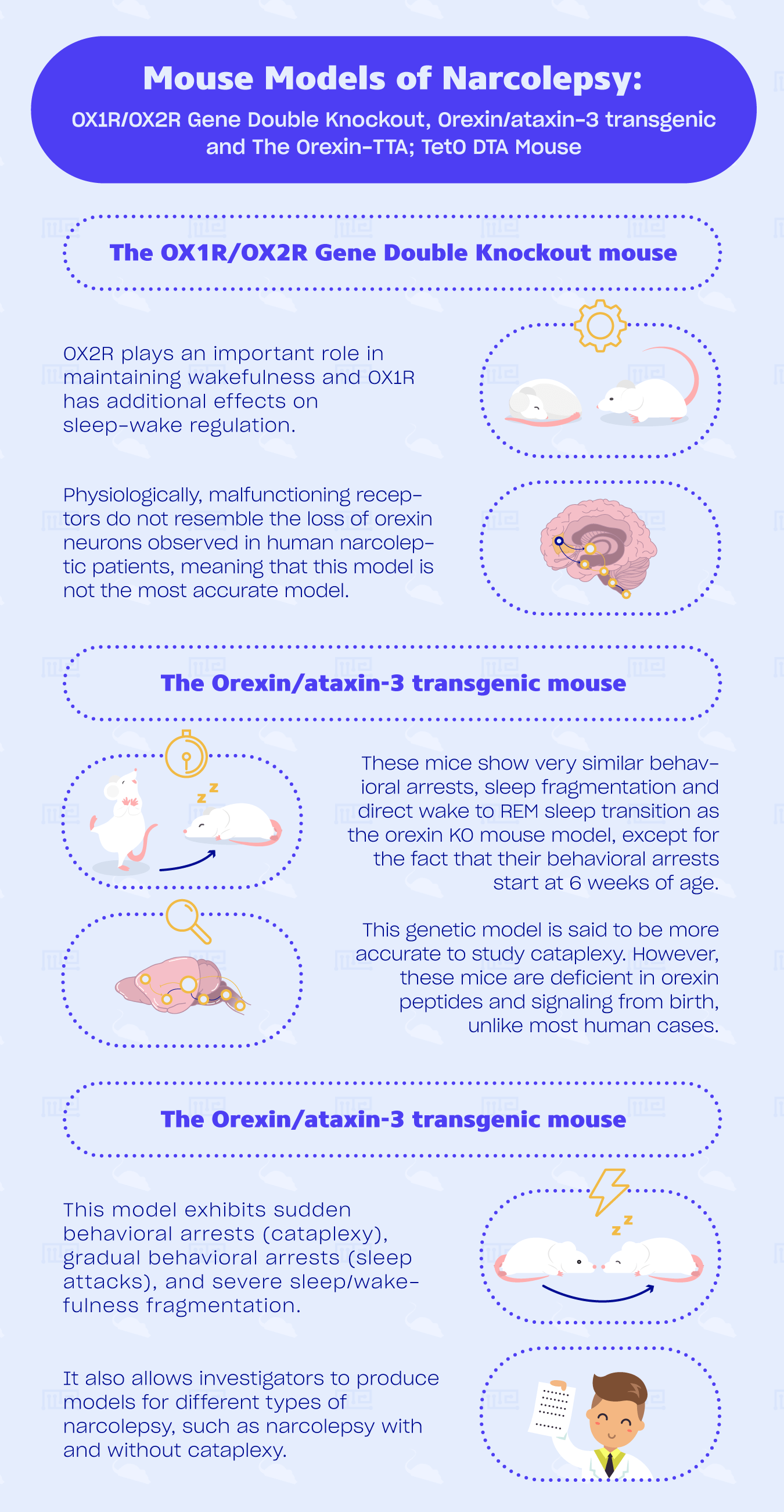
The OX1R/OX2R Gene Double Knockout mouse
Orexin KO mice and double receptor-knockout mice have virtually the same phenotype. Knowing that and the fact that OX2R−/− mice are only mildly affected by cataplexy and direct transition to REM sleep from awake states, we can deduce that OX2R plays an important role in maintaining wakefulness and OX1R has additional effects on sleep-wake regulation. It seems that both receptors have a critical role in gating of REM sleep[4] and mediate sleep induction. [8]
Another way of inhibiting the expression of both orexin receptors is by use of transcriptional blocking sequences. This results in mice with severely fragmented sleep and the inability to maintain sustained periods of wakefulness during the dark period compared to wild type littermates. However, no apparent cataplexy is found. [1]
This model is interesting when studying the contribution of both receptors to sleep/wake regulations.[8] They are relatively simple to breed and show the same phenotype as orexin KO mice.[1] However, physiologically, malfunctioning receptors do not resemble the loss of orexin neurons observed in human narcoleptic patients, meaning that this model, similar to orexin KO model, is not the most accurate model.
The Orexin/ataxin-3 transgenic mouse
These mice underwent selective postnatal degeneration of orexinergic neurons. As narcolepsy is not only caused by an almost complete loss of orexin neurons but also loss of dynorphin and NARP signaling from orexin neurons, a model of selective loss of orexin neurons more accurately resembles human narcolepsy than just orexin gene KO alone. Orexin neuron ablation can be achieved by expressing a cytotoxic transgene, the N-terminal truncated cDNA for human ataxin-3, in orexin neuron.[1][6] Read Hara et al. (2001) for a more detailed description of the method.[6]
These mice show very similar behavioral arrests, sleep fragmentation and direct wake to REM sleep transition as the orexin KO mouse model, except for the fact that their behavioral arrests start at 6 weeks of age, while orexin−/− mice could start showing them earlier than 3 weeks of age.[1] In addition, similarly to human narcoleptic patients, late-onset obesity can be observed (dependent on their genetic background) from week 12 to 15, which could be caused by sleep fragmentation-induced suppression of activity and energy expenditure.[1][3]
Due to their cataplectic attacks similar to human’s and its successful mimicking of loss of orexin neurons, this genetic model is said to be more accurate to study cataplexy. However, these mice are deficient in orexin peptides and signaling from birth, unlike most human cases.[1][5]
The Orexin-TTA;TetO DTA mouse
By removal of doxycycline from the diet of adult orexin-tTA;TetO DTA mice, in which diphtheria toxin A (DTA) is expressed in orexin neurons under control of the Tet-off system, 80% of orexin cells degenerate after one week of tet-off condition. Read Tabuchi et al. (2014) for a more detailed description of the methods.[1][4]
This model exhibits sudden behavioral arrests (cataplexy), gradual behavioral arrests (sleep attacks), and severe sleep/wakefulness fragmentation. Sudden behavioral arrests, however, can be observed only 14 days after the removal of doxycycline when less than 5 % of the orexin neurons remain. Cataplexy frequency increases for at least 11 weeks after doxycycline removal. Cataplexy occurs depending on the number of orexin neurons ablated.[5] Studies have shown that body weight might increase without a change in food consumption, similar to human narcolepsy, orexin KO and orexin/ataxin-3 mice.[1][4]
Previously mentioned animal models have helped decode the role of orexin neurons in sleep/wakefulness regulation. However, they lack the orexin peptides, receptors, or neurons from birth and thus do not replicate the typical post-pubertal onset of this disorder in humans. This model is interesting for pharmacological studies of sleep/wake fragmentation or cataplexy. It also allows investigators to produce models for different types of narcolepsy, such as narcolepsy with and without cataplexy.[5] A disadvantage is that it is a relatively new model and few studies have used them, which makes replication a bit more difficult.
Concluding remarks
For behavioral research, the two most used mouse models are orexin KO mice and orexin/ataxin-3 mice due to the fact that both of them show the most human-like narcoleptic episodes. The former mentioned is easier to breed and the latter is a more reliable model, due to the fact that the release of certain neurotransmitters is inhibited.
Current mouse models have limited utility in the development of novel pharmacological treatments for narcolepsy because cataplexy events are relatively infrequent. A good translational model for human cataplexy is orexin-tTA;TetO DTA mice as it ablates orexin neurons, the release of relevant neurotransmitters and allows for studying the aging of orexin deficiency and narcoleptic symptoms. However, the frequency of cataplectic episodes depends on the number of neurons ablated.
References
- Chen, L., Brown, R. E., Mckenna, J. T. & Mccarley, R. W. Animal Models of Narcolepsy. 296–308 (2009).
- Chemelli, R. M. et al. Narcolepsy in orexin Knockout Mice : Molecular Genetics of Sleep Regulation at Dallas. 98, 437–451 (1999).
- Toth, L. A. & Bhargava, P. Animal Models of Sleep Disorders. 63, 91–104 (2013).
- Sakurai, T. Animal Models of Narcolepsy. doi:10.1007/978-3-319-23078-8
- Tabuchi, S. et al. Conditional Ablation of Orexin / Hypocretin Neurons : A New Mouse Model for the Study of Narcolepsy and Orexin System Function. 34, 6495–6509 (2014).
- Hara, J. et al. Genetic Ablation of Orexin Neurons in Mice Results in Narcolepsy , Hypophagia , and Obesity Southwestern Medical Center at Dallas. 30, 345–354 (2001).
- Abbas, G., Shoji, H., Soya, S., Hondo, M. & Miyakawa, T. Comprehensive BehavioralAnalysis of Male Ox1r − / − Mice Showed Implication of Orexin Receptor-1 in Mood , Anxiety , and Social Behavior. 9, 1–10 (2015).
- Mang, G. M. et al. The Dual Orexin Receptor Antagonist Almorexant Induces Sleepand Decreases Orexin-Induced Locomotion by Blocking Orexin 2 Receptors.
- Willie, J. T. et al. Distinct Narcolepsy Syndromes in Orexin Receptor-2 and Orexin Null Mice : Molecular Genetic Dissection of Non-REM and REM Sleep Regulatory Processes Medical Center at Dallas. 38, 715–730 (2003).